鋰離子電池陰離子氧化還原化學(xué)的內(nèi)在科學(xué)及實用性討論
陰離子(X)氧化還原反應(yīng)的出現(xiàn),標(biāo)志著設(shè)計正極材料的思路發(fā)生了轉(zhuǎn)化型的變革——將近兩倍容量的增長。但是,伴隨著其發(fā)展的是眾多的溯源型基礎(chǔ)問題以及產(chǎn)業(yè)應(yīng)用上的全能考量。這篇綜述里,作者會探討其中一種可逆的、穩(wěn)定的陰離子氧化還原行為,并闡釋內(nèi)在機理。進一步地,關(guān)于實業(yè)應(yīng)用的限制以及其可行的解決方案也會被條分縷析地羅列以飼讀者。另外,作者也專門比對了這類材料與現(xiàn)行主流的高鎳系氧化物在市場上正面硬剛的戰(zhàn)場空間與勝率。
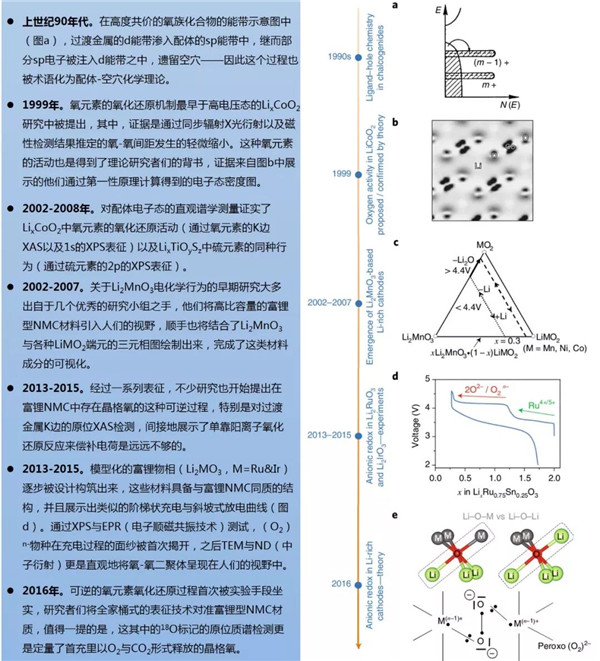
圖1陰離子氧化還原化學(xué)理論出爐的幾個關(guān)鍵節(jié)點
陰離子氧化還原化學(xué)的內(nèi)在科學(xué)(原理)
在嵌入式復(fù)合物的能帶結(jié)構(gòu)中,費米能級EF可以與電化學(xué)氧化還原(以下簡稱氧/還)電勢關(guān)聯(lián)起來,在這個結(jié)構(gòu)中,費米能級以上的空穴與費米能級以下的電子可以形成一個氧/還電對。含鋰過渡金屬氧化物的能帶結(jié)構(gòu)理論中只考慮到過渡金屬d軌道與氧元素的p軌道的重疊效應(yīng),這個現(xiàn)象直接導(dǎo)致了成鍵(M-O)與反鍵(M-O)*能帶的形成,前者具有強配體特征而后者具備金屬特性(圖1a-c)。(M-O)與(M-O)*能量之差也被稱為電荷轉(zhuǎn)移項Δ,其與M和O之間的電負(fù)性之差Δχ相關(guān)。電荷轉(zhuǎn)移項反映了M-O鍵的游離/共價特性:例如,將O替換成電負(fù)性更低的S之后,Δ降低(離子性更低),并且這個趨勢持續(xù)著從S直到Te。對于經(jīng)典的陰極材料來說,它們的氧/還過程只涉及到具有強烈金屬特性的(M-O)*能帶,因此也常被稱作陽離子氧化還原(過程)。
近來,理論學(xué)家們通過簡單的路易斯描述以及意識到富鋰材料能帶結(jié)構(gòu)中非鍵氧態(tài)的存在。這個路易斯排布中,O2-含有一個2s以及三個2p的二重態(tài),前者深埋在能級之中不具有氧化還原的活性。相反,高能氧的2p二重態(tài)參與到了M-O的成鍵過程,參與的程度與能帶結(jié)構(gòu)有關(guān)。不像更高O/M比的富鋰Li2MO3材料那樣,在經(jīng)典的層狀LiMO2(O/M=2,圖1d)中所有的這三個2p軌道都參與到了M-O成鍵階段。可以肯定的是,在類Li2MnO3型結(jié)構(gòu)中,氧2p軌道中的一個電子(指向Li1/3M2/3O2層內(nèi)鋰的那一個)的鍵合比較虛弱,原因是它與鋰2s軌道的重疊程度相對較小。因此,它就像一個氧的非鍵態(tài)那樣,并固定在穩(wěn)定了的(M-O)成鍵能帶的上方(圖1c)。特別提一下,這個電子也有時被稱作氧的2p“孤”或者“未雜化”態(tài),或者“O的孤對電子”,抑或“鋰-氧-鋰”構(gòu)型,又或者C2v點群對稱中的“b1*態(tài)”—— 導(dǎo)致了語義上不必要的困惑。
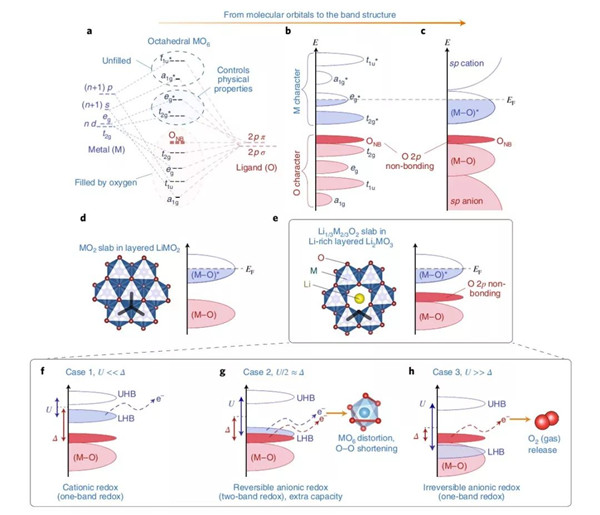
圖2. 從分子軌道到能帶結(jié)構(gòu)細(xì)說M-O配位理論
為什么這個氧的非鍵態(tài)在電化學(xué)上如此吸睛?這是因為,與在傳統(tǒng)體系中一旦(M-O)*被掏空電子就只能取自于本已穩(wěn)定的(M-O)成鍵能帶這一變化不同,這個非鍵態(tài)可以作為除了常用(M-O)*能帶之外第二個提供額外電子的能帶,在不罹患結(jié)構(gòu)失穩(wěn)的前提下為容量提升添磚加瓦。而能夠觸發(fā)這雙能帶氧/還過程的關(guān)鍵依賴于(M-O)*反鍵能帶與氧2p非鍵能帶的相對位置。
為了更好的理解能帶的占位現(xiàn)象,本文作者首先需要引入d-d庫倫相互作用量U,而這并不是一個電池化學(xué)家經(jīng)常引述的明確概念,反而是頻頻被固體物理學(xué)家用來表征d軌道場內(nèi)電子斥力的術(shù)語。這個物理量更接近于描述場內(nèi)的定域電子而不是動能,它分裂了部分占據(jù)的(M-O)*能帶(被叫做莫特-哈伯德分裂),形成了空置的上哈伯德能帶與滿電子的低哈伯德能帶(在圖2f中分別被叫做UHB與LHB)。更精確的講,U反比于軌道體積,因此它強相關(guān)于參與進來的d金屬。所以,它伴隨周期表從左至右的軌道收縮而增加,伴隨3d至5d過渡金屬的軌道膨脹而降低。
LHB位置與氧2p非鍵能帶的相對位置依賴于U對Δ的相對值,這也衍生出三種不同的情形(圖2f-h)。首先,對于U<<Δ的情形,其適用于大量的氧化物與氟化物,這些物質(zhì)具備高度離子化的M-L鍵(L即配體),電子也取自于滿態(tài)LHB(圖2f)之中,就如經(jīng)典的單能帶陽離子氧化還原情形一樣。然后,便是與第一種情況正相反的U>>Δ(圖2h),在這種狀況下,單能帶的氧化還原依舊主導(dǎo),只不過電子是直接取自于居滿態(tài)LHB之上氧2p的非鍵能帶。得益于定域氧2p非鍵態(tài)的強化學(xué)硬度,這種條件會制造出高度活潑的On-物種,這類物質(zhì)可能會通過還原消除或者攻擊電解液而游離出金屬網(wǎng)絡(luò)。這便導(dǎo)致了常見的Li2MnO3、相關(guān)的富鋰NMC或者Li4FeTeO6以及Li4FeSbO6陰極中的部分不可逆過程。對于富鋰NMC材料來說,如前述報道的附近Mn4+陽離子能在多大程度上穩(wěn)定On-物種?答案是無法完全穩(wěn)定,因為隨著這類材料爬上首周充電的平臺階段,氧流失的現(xiàn)象始終能被觀測到。值得注意的是,這種氧與鋰的同時脫出顯著地改變了富鋰NMC材料的初始能帶結(jié)構(gòu),在之后的過程中它可能會變得更加傾向于可逆的陰離子氧化還原反應(yīng)以及O-O二聚體的形成。當(dāng)然,捕捉這種能帶結(jié)構(gòu)動力學(xué)的變化需要進一步的DFT研究。
最后,U/2≈Δ這種中間情形(圖2g),會導(dǎo)致LHB與氧2p非鍵能帶的重疊,這樣的條件對于階段式(Li2-xRuO3)與連續(xù)式(Li2-xIrO3)的電化學(xué)過程都十分有利,因而可以獲得雙倍的容量。在這種情況下,電子的轉(zhuǎn)移會導(dǎo)致費米能級的簡并,使其失穩(wěn)。為了規(guī)避這種不穩(wěn),簡并度一般會通過姜-泰勒或者皮爾斯畸變提升,這種效應(yīng)導(dǎo)致氧框架重組、對稱性的降低來縮短某些O-O間距確保穩(wěn)定化的M-(O2)n-相互作用的發(fā)生。這種過氧類O-O二聚體的穩(wěn)定化作用之前被稱作“還原性偶聯(lián)”,與配位化學(xué)頗為相似。以上敘述解釋了實驗上觀察到的Li2IrO3與Li2RuO3中出現(xiàn)的MO6八面體扭曲現(xiàn)象。于是有了一種獨特的現(xiàn)象:電子部分地從陰離子的非鍵能帶中脫出,因此把之稱為陰離子氧化還原。這與高度脫鋰態(tài)的LixCoO2全然不同,經(jīng)Bader電荷計算、XAS與XPS分析表明,高度脫鋰LixCoO2中氧是具有氧/還活性的,這是因為其高度的共價性將顯著的氧特征傳達給了氧/還活性的(M-O)*能帶。作者們因此謹(jǐn)慎地反對把這種情況稱之為“陰離子氧化還原反應(yīng)”,因為它基本上也只是一個單能帶過程,并不提供額外的容量。
綜上,依據(jù)這些能帶圖表可知,要想獲得多余的容量,U/2≈Δ是必要的,而這也為材料設(shè)計師們打開了大門:通過適當(dāng)篩選金屬-配體的組合來達到Δ與U的微妙平衡。相反地,研究者們應(yīng)該盡量避免的是不可逆氧流失的情形。理論學(xué)家們通過計算3d、4d以及5d金屬與氧配位時不同含鋰量下氧流失反應(yīng)的焓變(LixMO3→LixMO3-δ+δ/2 O2)。計算的結(jié)果突顯了在3d金屬配位氧體系中要避免氧流失情形的難度要比4d與5d金屬中難得多。考慮到原材料成本之后,這個理論計算的結(jié)果盡顯悲觀,但即便如此,也不應(yīng)該放棄3d金屬配位氧體系,目前已經(jīng)有不少工藝方面的改進策略在減緩富鋰NMC材料的氧流失。
總體上看,無論是強共價性(配位)還是配體中的非鍵p能帶,單打獨斗的情況之下都無法滿足可逆氧化還原反應(yīng)進行的條件。例如,只談共價性,盡管早期對于TiS2的XAS檢測表明了S元素具有強力的氧化還原作用,但也僅僅是因為高度共價性的反鍵(M-O)*態(tài)具有明顯的配體特征導(dǎo)致的,但這個過程并沒有產(chǎn)生額外的容量,至少并沒有通過所認(rèn)知的陰離子氧化還原過程釋放容量。同樣的,對于氧的非鍵2p態(tài)來說,它們也可以存在于聚陰離子型的化合物中,比如LiFePO4或者LiFeSO4F,但這類離子型化合物因為非鍵2p態(tài)深埋在費米能級之下因而無法催生具備活性的陰離子氧化還原反應(yīng),所以它們對外表現(xiàn)為陽離子型的氧化還原機制。
盡管上述場景是建立在氧化物之中,但你也可以將早期的工作推衍至整個氧族化合物之中。比如,在三硫化物TiS3之中,非原位XPS表征的結(jié)果清晰明了:鋰在電化學(xué)回嵌過程中先伴隨著S-S二聚體的消失[(S2)2-+2e-→2S2-],然后是Ti4+/3+的還原。簡單講,TiS3中的Ti4+并不能滿足S2-態(tài)(Ti6+是不可能的),因此引發(fā)結(jié)構(gòu)畸變來重新混合空置的硫的3p非鍵能級以及形成(S2)2-二聚體可以最終穩(wěn)定TiS2-(S2)2-結(jié)構(gòu),并且(S2)2-還能如上所述進行電化學(xué)還原反應(yīng)。如果記憶力好一些,你會發(fā)現(xiàn)這種陰離子與陰離子的相互作用可以追溯到二聚體之外的一種強共價晶格的物質(zhì)比如IrTe2[或者說Ir3+(Te3/2-)2],它展示出一種Te-Te亞晶格的聚合來形成所謂的聚CdI2型結(jié)構(gòu)。不過,盡管這種借助全滿非鍵配位能態(tài)理論的集大成解釋在描述富鋰氧化物及相關(guān)氧族化合物上令人滿意,但在那些新發(fā)現(xiàn)的材料上,尤其是針對異常特征它還是會持續(xù)受到挑戰(zhàn)的。特別指出,同樣具備氧非鍵2p態(tài)(指向鎂)的貧鈉型Na2/3[Mg0.28Mn0.72]O2,之前關(guān)于其陰離子氧/還機制的報道結(jié)果都基本上復(fù)合前述的理論體系范疇。不過,最近在相關(guān)報道中,氧活動出現(xiàn)在了具有類似結(jié)構(gòu)但MO2層內(nèi)并不含堿金屬也無堿土金屬的化合物之中,這就很讓人起興:這種物質(zhì)是明顯不具有氧的非鍵2p能態(tài),真相如何還有待進一步探索。當(dāng)然,除了解釋陰離子氧化還原機制之外,這套理論同樣也為新型高比容量的材料搜索提供指導(dǎo)意見。
陰離子氧/還機制與富鋰陰極材料的實用性(討論)
在見證了陰離子氧/還反應(yīng)的高容量潛質(zhì)如何激發(fā)成分與結(jié)構(gòu)多樣化的新材料設(shè)計之后,作者接下來會評估它們在實際應(yīng)用中的可操作性,于此你會發(fā)現(xiàn)高比容量只不過是幾個迫切需求的其中之一。除了具備更高的比容量之外,一個新材料也必須在倍率性能、能量效率與循環(huán)穩(wěn)定性上超越現(xiàn)有的,并且還要在成本與安全上保持競爭力。此外,還需要先進的電化學(xué)測試手段(百寶箱3)來專門定睛于與陰離子氧/還型化合物密切相關(guān)的特定電化學(xué)性質(zhì)。

圖3評估新型陰離子氧/還陰極材料實用性的正確打開方式
以常見的富鋰NMC材料開始。盡管經(jīng)歷了數(shù)年學(xué)術(shù)與工業(yè)界的努力,這類材料還依舊在等待商業(yè)化的春天。從第一周中就可以針砭出實用化的問題,它那特別的兩步化第一周充電曲線以一個典型的脫嵌型陽離子氧/還階段開始,這一過程沒毛病,接著就是一個陰離子氧/還階段(直線區(qū)域)并終結(jié)于不可逆的產(chǎn)氣。除了可以制造出明顯破壞電池壽命的寄生反應(yīng)之外,這種不可逆問題同樣會導(dǎo)致全電池的過平衡(更重)。而且,陰離子氧化會永久的改動電化學(xué),并形成S型的充放電曲線。一旦直線區(qū)域穩(wěn)定下來,這種結(jié)合了陽離子與陰離子氧/還的活性會提供超高的容量,即便如此,體系仍然會出現(xiàn)大幅度的電壓遲滯(>400 mV),從而折損能量效率。這種遲滯源自動力學(xué)而不是動力學(xué)——也就是說,即便電流接近零也不會消除——并會導(dǎo)致充電vs放電dQ/dV曲線的不對稱。它最終會引發(fā)路勁依賴,進而使得充電深度(SoC)管理變得十分復(fù)雜。由于遲滯減損了能量效率并引發(fā)周次的能量損耗(假設(shè)以熱量形式散發(fā)),它還會成為一個成本難題,尤其對諸如電動汽車以及電站等大型應(yīng)用而言。所以,這個特征(問題)即不容富鋰NMC材料忽略,也為過分炒作的那些轉(zhuǎn)化型陽極蓋棺定論了。
只有寥寥研究嘗試去理解遲滯現(xiàn)象的本質(zhì)。在這些工作中,電壓窗口實驗可以展示高電壓的陰離子氧化對應(yīng)于其在低電壓的還原情況。核磁共振測試結(jié)果輔以相變和格子氣模型,認(rèn)為不可逆陽離子遷移與電壓遲滯相關(guān)。尤其是在長循環(huán)下,這種關(guān)聯(lián)是否暗示因果關(guān)系目前尚待商榷。有趣的是,一項最近的研究同樣指出了陰離子氧/還反應(yīng)與陽離子遷移間的相關(guān)性,宣稱這兩種效應(yīng)是相互耦合的。
不過,人們也必須指導(dǎo)陽離子遷移實際是是陰離子氧化之后結(jié)構(gòu)失穩(wěn)的結(jié)果。順著這個思路,近來組合光譜技術(shù)(HAXPES和XAS)與電化學(xué)表征手段展示了陰離子氧化還原與電壓遲滯的直接關(guān)聯(lián)。通過關(guān)聯(lián)富鋰NMC材料的電化學(xué)阻抗與其電荷補償機制,研究者們還發(fā)現(xiàn)與陽離子那快速的氧/還過程相反,陰離子氧/還在高低電壓下俱受制于遲緩的動力學(xué)過程。電壓遲滯再加上動力學(xué)遲緩的結(jié)果就是,富鋰陰極材料的總體極化變大。現(xiàn)今,一個揮之不去的基礎(chǔ)問題是,為什么陰離子氧/還反應(yīng)會與電壓遲滯與慢速動力學(xué)過程相關(guān),這個問題目前仍需理論上的深入研究。一個合理的假設(shè)是能量損耗(因此慢速)的短程原子移動關(guān)聯(lián)到過渡金屬持續(xù)的、有陰離子氧/還驅(qū)動的遷移以及充放電中氧-氧二聚體的持續(xù)形成/斷裂。因此,氧元素框架的結(jié)構(gòu)彈性可以被認(rèn)為是一個應(yīng)對這些問題的關(guān)鍵條件。
在實驗方面,盡管歷經(jīng)了大量化學(xué)方法的嘗試,但富鋰NMC中的遲滯問題仍然沒有得到解決。有趣的是,在4d金屬配位氧型的富鋰Li2Ru0.75Sn0.25O3(LRSO)材料中,這個缺陷得到了一定的控制(~200 mV),在這種氧化物中,遲滯現(xiàn)象明顯是在陰離子氧化還原反應(yīng)的高電壓階段被觸發(fā)的。而且,LRSO中也出現(xiàn)了類似富鋰NMC中的dQ/dV曲線不對稱現(xiàn)象,但是程度明顯要輕。通過對dQ/dV曲線中陽離子-陰離子過程的去耦化,原位XAS檢測直觀地表明這種不對稱現(xiàn)象源于陰離子的氧化還原反應(yīng)。除了電壓遲滯之外,LRSO模型系統(tǒng)也幫助闡釋了慢速陰離子動力學(xué)過程。綜上,在富鋰NMC(3d金屬型應(yīng)用體系)與LRSO(4d金屬型模型體系)中都有相似的發(fā)現(xiàn),即陰離子氧化還原反應(yīng)在引發(fā)電壓遲滯與慢速動力學(xué)過充中產(chǎn)生了有害影響,這個發(fā)現(xiàn)引人深思,因為其他陰離子氧/還型陰極也出現(xiàn)相似或者更明顯的缺陷。舉個例子,頗具潛力的低成本、高容量型巖鹽Li1.2Mn0.4Ti0.4O2即便在50℃下還是出現(xiàn)了充電與放電電壓的巨大差值,不得不讓人聯(lián)想到慢速動力學(xué)與電壓遲滯。唯一例外于這些缺陷的是Li2IrO3多態(tài)物,它們展示出的是疊加型充放電曲線。言而總之,陽離子與陰離子氧化還原反應(yīng)間的相互作用依賴于化學(xué)組成與共價程度,它直接影響了富鋰材料中那些及其重要的實用性質(zhì),比如動力學(xué)與遲滯。

圖4富鋰材料的現(xiàn)實難題
與這兩個相對較新的概念不同,在富鋰NMC材料早期研究中便已確認(rèn)的缺點:電壓衰減,已經(jīng)廣受調(diào)研,它會逐漸地降低能量輸出并使得SoC管理復(fù)雜化。現(xiàn)在已經(jīng)可以明確的是,在高電壓下陰離子具有氧化還原活性的富鋰NMC材料中,電壓衰減明顯加劇,這個現(xiàn)象在LRSO模型系統(tǒng)中也得到了進一步的證實。而且,LRSO的研究幫助開發(fā)了一種錫穩(wěn)定型的Li2RuO3改善方案,使得電壓衰減在與鈦摻雜體系相比之下得到明顯的抑制(圖4d)。一種合理的解釋是,隨著長循環(huán)的進行,四面體位點逐漸囚困住了小尺寸的鈦離子。這種推理同樣適用于富鋰NMC材料的電壓衰減,在高電壓下小尺寸的錳離子在氧流失的協(xié)助下促進了“層狀-尖晶石”的漸變。在其他綜述中列出的多種化學(xué)方案,從表面改性到化學(xué)取代再到電解液添加劑,無一不都是旨在解決富鋰NMC材料中的電壓衰減。某些方法確實減緩了電壓衰減,但完全根除這個缺陷貌似不可能。
最后,以獲取更多的陰離子容量高電壓充電策略最終會引發(fā)氧釋放,并惡化材料的穩(wěn)定性。這種高活性與欠穩(wěn)定之間的強相關(guān)并不陌生,在其他電化學(xué)體系中比如水分解催化劑屢見不鮮。在富鋰NMC材料首周充電階段出現(xiàn)的陰離子活化平臺是結(jié)構(gòu)失穩(wěn)的一個早期指標(biāo),這個平臺反應(yīng)所釋放的晶格氧被認(rèn)為會持續(xù)出現(xiàn)在后續(xù)循環(huán)中,只不過因為阻氧表面的重構(gòu)會降低晶格氧釋放的速率。阻氧現(xiàn)象也為粒子級設(shè)計方案提供了一種動力:保護材料的體相不受氧釋放的威脅,這些方案就包括了核殼或者濃度-梯度顆粒的合成。順便一提,即便是無序巖鹽相也無法在長循環(huán)下保持穩(wěn)定。目前,最具潛力的體系是β-Li2IrO3模型,這種可以支撐鋰完全脫出的物相突顯了三維結(jié)構(gòu)的重要程度,不失為未來開發(fā)穩(wěn)定的陰離子氧/還陰極材料道路上的一盞明燈。
總的來說,富鋰材料的實際可行性緊密依靠于陰離子氧/還過程,基于此,改良方案必須直接指向它。這類陰極的缺陷并不會陷它們與萬劫不復(fù),問題只是在于如何將合適的陰極匹配進正確的體系與應(yīng)用。
官方微信售電那點事兒")
責(zé)任編輯:繼電保護
- 相關(guān)閱讀
- 泛在電力物聯(lián)網(wǎng)
- 電動汽車
- 儲能技術(shù)
- 智能電網(wǎng)
- 電力通信
- 電力軟件
- 高壓技術(shù)
-

權(quán)威發(fā)布 | 新能源汽車產(chǎn)業(yè)頂層設(shè)計落地:鼓勵“光儲充放”,有序推進氫燃料供給體系建設(shè)
2020-11-03新能源,汽車,產(chǎn)業(yè),設(shè)計 -

中國自主研制的“人造太陽”重力支撐設(shè)備正式啟運
2020-09-14核聚變,ITER,核電 -

探索 | 既耗能又可供能的數(shù)據(jù)中心 打造融合型綜合能源系統(tǒng)
2020-06-16綜合能源服務(wù),新能源消納,能源互聯(lián)網(wǎng)
-
新基建助推 數(shù)據(jù)中心建設(shè)將迎爆發(fā)期
2020-06-16數(shù)據(jù)中心,能源互聯(lián)網(wǎng),電力新基建 -

泛在電力物聯(lián)網(wǎng)建設(shè)下看電網(wǎng)企業(yè)數(shù)據(jù)變現(xiàn)之路
2019-11-12泛在電力物聯(lián)網(wǎng) -

泛在電力物聯(lián)網(wǎng)建設(shè)典型實踐案例
2019-10-15泛在電力物聯(lián)網(wǎng)案例
-

新基建之充電樁“火”了 想進這個行業(yè)要“心里有底”
2020-06-16充電樁,充電基礎(chǔ)設(shè)施,電力新基建 -

燃料電池汽車駛?cè)雽こ0傩占疫€要多久?
-

備戰(zhàn)全面電動化 多部委及央企“定調(diào)”充電樁配套節(jié)奏
-
權(quán)威發(fā)布 | 新能源汽車產(chǎn)業(yè)頂層設(shè)計落地:鼓勵“光儲充放”,有序推進氫燃料供給體系建設(shè)
2020-11-03新能源,汽車,產(chǎn)業(yè),設(shè)計 -
中國自主研制的“人造太陽”重力支撐設(shè)備正式啟運
2020-09-14核聚變,ITER,核電 -

能源革命和電改政策紅利將長期助力儲能行業(yè)發(fā)展
-
探索 | 既耗能又可供能的數(shù)據(jù)中心 打造融合型綜合能源系統(tǒng)
2020-06-16綜合能源服務(wù),新能源消納,能源互聯(lián)網(wǎng) -

5G新基建助力智能電網(wǎng)發(fā)展
2020-06-125G,智能電網(wǎng),配電網(wǎng) -

從智能電網(wǎng)到智能城市